Cell-人腸道微生物廣泛的菌株水平的拷貝數(shù)變異
2015-10-30
人體腸道微生物在宿主代謝、免疫和藥物反應(yīng)中扮演重要角色,與人類健康息息相關(guān),然而每個(gè)菌種包含很多不同菌株,這些菌株編碼的基因和每個(gè)基因的拷貝數(shù)可能差別很大,這種種內(nèi)變異賦予不同菌株潛在的功能差異,因此不能單從菌種組成推斷菌群功能,物種水平的比較分析可能無(wú)法獲得樣品間重要的功能差異。
Sharon Greenblum等人以之前研究中的109個(gè)健康、肥胖、炎癥性腸病(IBD)個(gè)體的腸道微生物宏基因組樣本為實(shí)驗(yàn)對(duì)象,運(yùn)用新建立的分析方法直接從109個(gè)樣品的宏基因組數(shù)據(jù)估算給定樣本中的微生物菌種的每個(gè)基因的拷貝數(shù),進(jìn)而發(fā)現(xiàn)樣本間的拷貝數(shù)變異。研究者總共將超過(guò)24.5 億條的 75 bp 短序列map到 235個(gè)參考基因組(分為96 個(gè)genome clusters)上,運(yùn)用建立的分析方法估計(jì)了幾千個(gè)KOs的拷貝數(shù),并利用獲得的拷貝數(shù)估計(jì)的數(shù)據(jù)集,探索了腸道這一高度復(fù)雜的生態(tài)系統(tǒng)中的中性和適應(yīng)性的變異,將宏基因組水平基因豐度的不同與基因組水平的變異聯(lián)系起來(lái)。
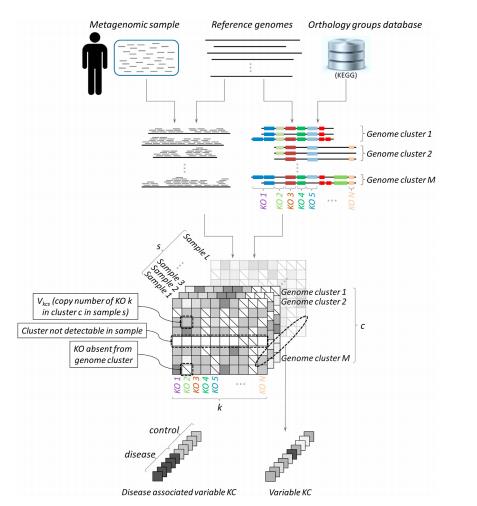
分析方法示意圖
研究發(fā)現(xiàn),拷貝數(shù)變異在腸道環(huán)境十分普遍,有的物種超過(guò)20%的基因都有顯著的拷貝數(shù)變異,這些變異或許可以解釋之前很多研究中出現(xiàn)的物種水平變化趨勢(shì)和基因水平變化趨勢(shì)存在差異的現(xiàn)象。通過(guò)分析研究者共檢測(cè)到38個(gè)genome cluster的735個(gè)highly variable KCs,并檢測(cè)到40個(gè) genome clusters的 5,004個(gè)set-specific variable KCs。研究者發(fā)現(xiàn)每個(gè)cluster的highly variable KCs數(shù)變化很大,例如在Roseburia intestinalis cluster中多達(dá)47個(gè)(占此cluster全部KCs的4.05%),而平均每個(gè)cluster的highly variable KCs占1.79%。此外,很多反映適應(yīng)性的變異為set-specific variable KCs,變異不顯著且僅在少量樣本中出現(xiàn)。
研究者將每個(gè)cluster的variable KCs與cluster中已測(cè)序的參考基因組的已知變異進(jìn)行比較發(fā)現(xiàn),數(shù)據(jù)庫(kù)中的參考基因組并未包含全部的種內(nèi)變異,同樣,研究所用的樣本可能也不包含很多參考基因組中的變異。
本研究發(fā)現(xiàn)運(yùn)輸功能相關(guān)的KCs非常傾向于高拷貝數(shù)變異,即成為highly variable KCs,相當(dāng)大比例的腸道微生物物種中運(yùn)輸功能的基因拷貝數(shù)存在靈活性,特定的運(yùn)輸功能的基因可能會(huì)促進(jìn)微生物對(duì)腸道環(huán)境的適應(yīng)性。此外,大多數(shù)set-specific variable KCs似乎與物種對(duì)環(huán)境的反應(yīng)和與環(huán)境的相互作用有關(guān),顯示了腸道菌群強(qiáng)大的適應(yīng)性潛能。研究結(jié)果顯示,盡管變異是可能是個(gè)體性的,但某些(如與環(huán)境適應(yīng)性相關(guān)的)基因和功能可能普遍傾向于發(fā)生變異。
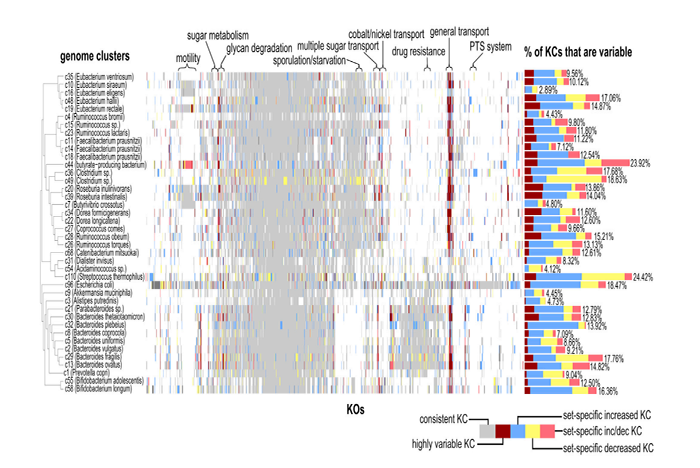
Variable KCs示意圖
一些變異可能表現(xiàn)了對(duì)特定宿主表型的適應(yīng)。研究者對(duì)在健康個(gè)體和肥胖或IBD個(gè)體間顯著不同的可變KCs進(jìn)行研究,最終發(fā)現(xiàn)24個(gè)KCs的拷貝數(shù)與IBD顯著相關(guān),3個(gè)KCs的拷貝數(shù)與肥胖顯著相關(guān)(FDR<0.05)。該發(fā)現(xiàn)進(jìn)一步揭示了單物種對(duì)疾病的作用,例如, Roseburia inulinivorans中編碼主要藥物外排蛋白的基因(K08217)的拷貝數(shù)的增加與IBD久治不愈高度相關(guān)。
研究者使用回歸分析將每個(gè)cluster的拷貝數(shù)去卷積成為數(shù)據(jù)庫(kù)中所含菌株的線性組合。在有很多已測(cè)序基因組的cluster中,大多數(shù)樣本的拷貝數(shù)可以用已知菌株的線性組合表示;而在只有少數(shù)菌株基因組序列已知的cluster中,有時(shí)只有部分拷貝數(shù)變異可以被表示。為進(jìn)一步比較樣本間拷貝數(shù)變異情況,研究者使用了主成分分析,此分析揭示了每個(gè)cluster內(nèi)的復(fù)雜的群落結(jié)構(gòu),各樣本間差異明顯,表明個(gè)體化變異普遍存在。
本研究證明了種水平的研究導(dǎo)致的功能信息缺失的程度,有利于促進(jìn)菌株水平微生物群落結(jié)構(gòu)的研究,表明了分析微生物群落的種內(nèi)變異對(duì)理解物種組成與群落功能之間的復(fù)雜關(guān)系非常重要。此外,本研究采用建立的分析方法定量表征了腸道微生物的變異,獲得的數(shù)據(jù)集和研究方法可作為未來(lái)人類微生物或其他環(huán)境中微生物組成變化研究的寶貴資源。
本文由市場(chǎng)部技術(shù)支持 王紅整理




















