在上一期的“5分鐘小課堂”中,小編梳理了宏基因組學(xué)研究的關(guān)鍵步驟流程,并概述了挖掘宏基因組學(xué)大數(shù)據(jù)的四大“法寶”:拼接組裝、功能注釋、生物標(biāo)記物篩選和菌株水平的精細(xì)解析。其中,首當(dāng)其沖的就是宏基因組的拼接組裝,可謂高通量測序中的“拼圖游戲”。作為后續(xù)三大“法寶”的數(shù)據(jù)來源,拼接組裝的效果將直接影響下游分析的可靠性,對研究意義的重要性不言而喻。今天,就讓小編與您一起,探索宏基因組拼圖游戲的奧秘,獻(xiàn)上史上最全攻略!
1. 什么是拼接?
拼接組裝是根據(jù)序列的一致性,將高通量測序產(chǎn)生的眾多宏基因組DNA短片段依次有序地重疊連接在一起,從而“重建”獲得較長的連續(xù)不間斷序列,也就是傳說中的“Contigs”。利用雙端PE(Paired-end)序列攜帶的信息,可以估計Contigs之間的間隔長度(即Gaps),從而連接形成Scaffolds。
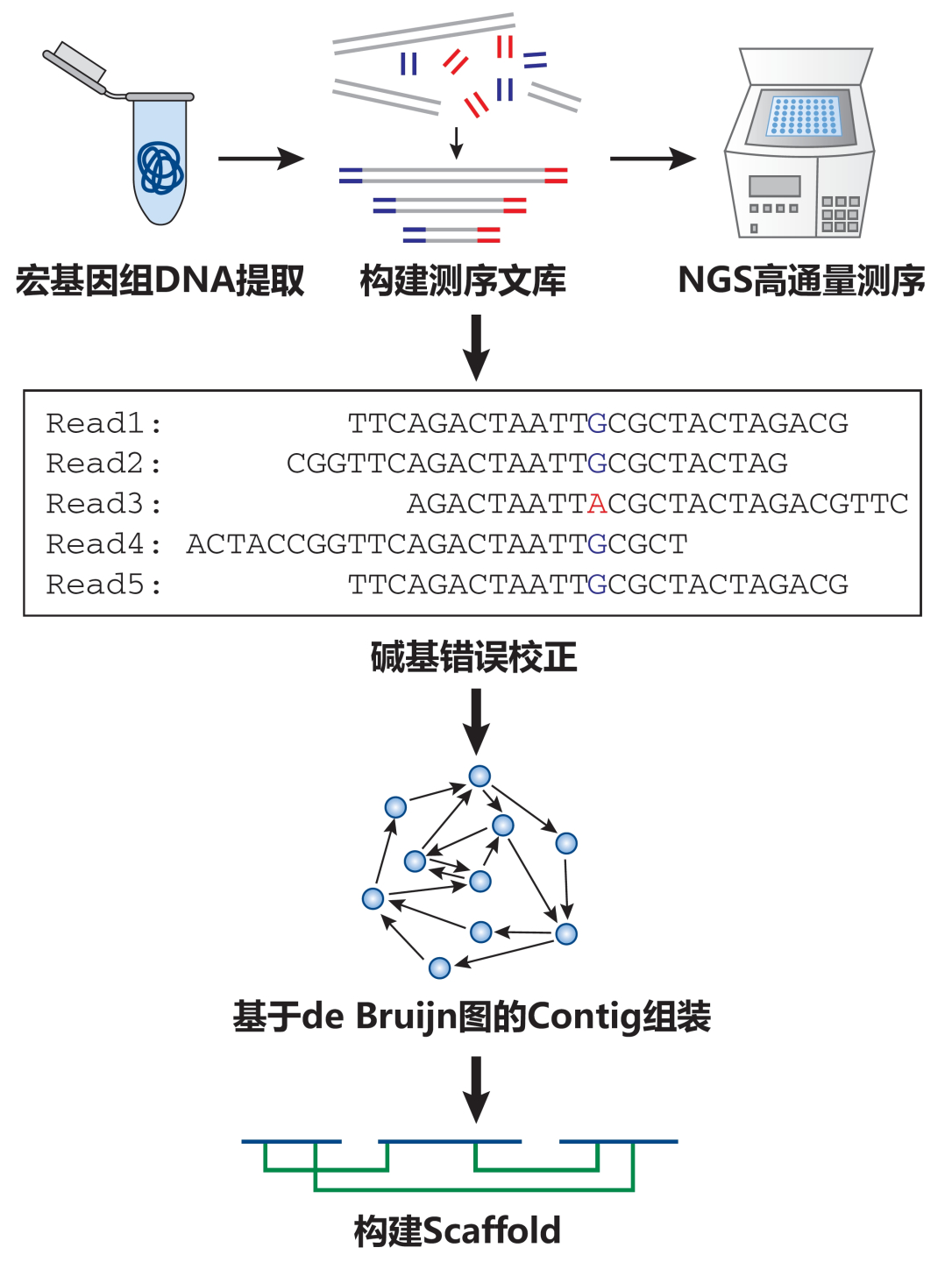
宏基因組的拼接組裝流程圖,修改自文獻(xiàn)[1]
通過以上介紹,小伙伴們有沒有發(fā)現(xiàn),宏基因組拼接組裝和拼圖游戲真的有異曲同工之妙呢!這里,拼圖的原材料就是短片段序列,拼出的圖就是Contigs和Scaffolds,而游戲的通關(guān)秘訣,無疑就是選取合適的拼接組裝算法啦!
2. “拼圖游戲”的關(guān)鍵:de Bruijn圖和一筆畫問題
宏基因組包含成千上萬種微生物,彼此之間的含量差異可達(dá)好幾個數(shù)量級。拼接組裝這樣的“大雜燴”,不僅需要龐大的數(shù)據(jù)量,更需要選取精巧、合適的算法。
目前的宏基因組大數(shù)據(jù)通常由Illumina HiSeq測序儀產(chǎn)生,序列較短(2 × 150 bp)但通量極高。因此,科學(xué)家對序列拼接組裝的算法做了針對性的優(yōu)化,將拼圖游戲簡化為我們熟知的“一筆畫問題”圖論問題,下圖就是個鮮活生動的例子:
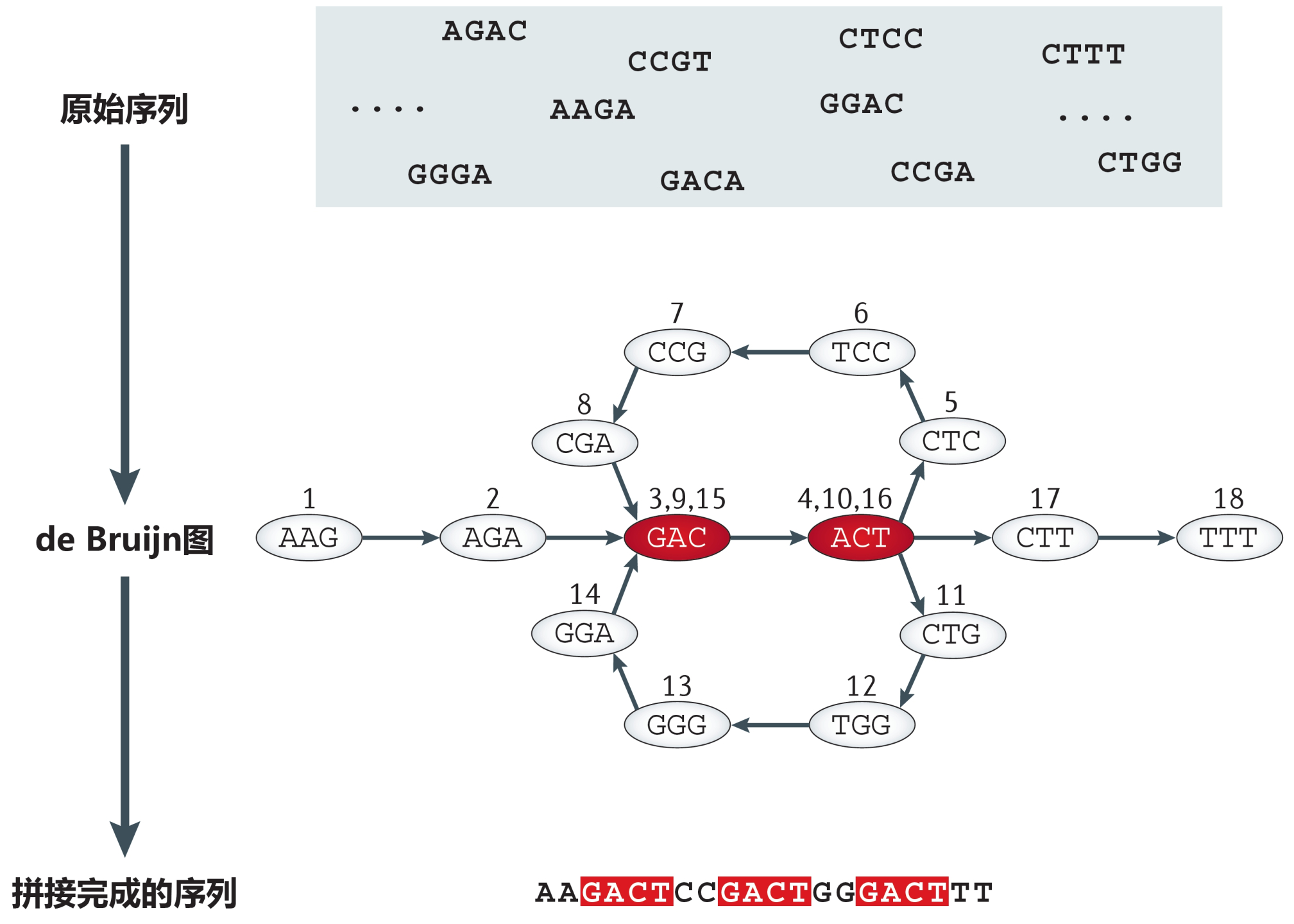
基于de Bruijn圖的序列拼接組裝示意圖,修改自文獻(xiàn)[2]
上圖中,原始序列長度為4堿基。首先將每一條短序列都拆分為一系列長度為k的子片段(俗稱k-mer),比如,圖中的原始序列AAGA被拆分為AAG和AGA兩個長度為3堿基的k-mer。然后根據(jù)全體k-mer之間的連接順序和重疊關(guān)系構(gòu)建de Bruijn圖,嘗試找到一次性遍歷所有k-mer的“一筆畫”路徑,由此完成拼接組裝,獲得Contigs和Scaffolds序列(圖中的紅色部分為重復(fù)序列,可以看到,它們也被正確識別和拼接)。
3. 常用拼接組裝工具簡介
目前,基于de Bruijn圖的序列拼接組裝工具已成為主流,包括SOAPdenovo2[3]和IDBA-UD(Iterative De Bruijn graph Assembler for sequencing data with highly Uneven Depth)[4]等。這些工具都能對原始序列中隱含的測序錯誤進(jìn)行校正,從而提升拼接組裝的精確度。通常而言,SOAPdenovo2的拼接速度較快,而IDBA-UD采用了迭代算法,從一系列k-mer值中,選取最合適的k-mer參數(shù)進(jìn)行拼接組裝,同時針對宏基因組中不同物種測序深度不均一的現(xiàn)象進(jìn)行了優(yōu)化,因而被認(rèn)為更適合于宏基因組的拼接組裝。
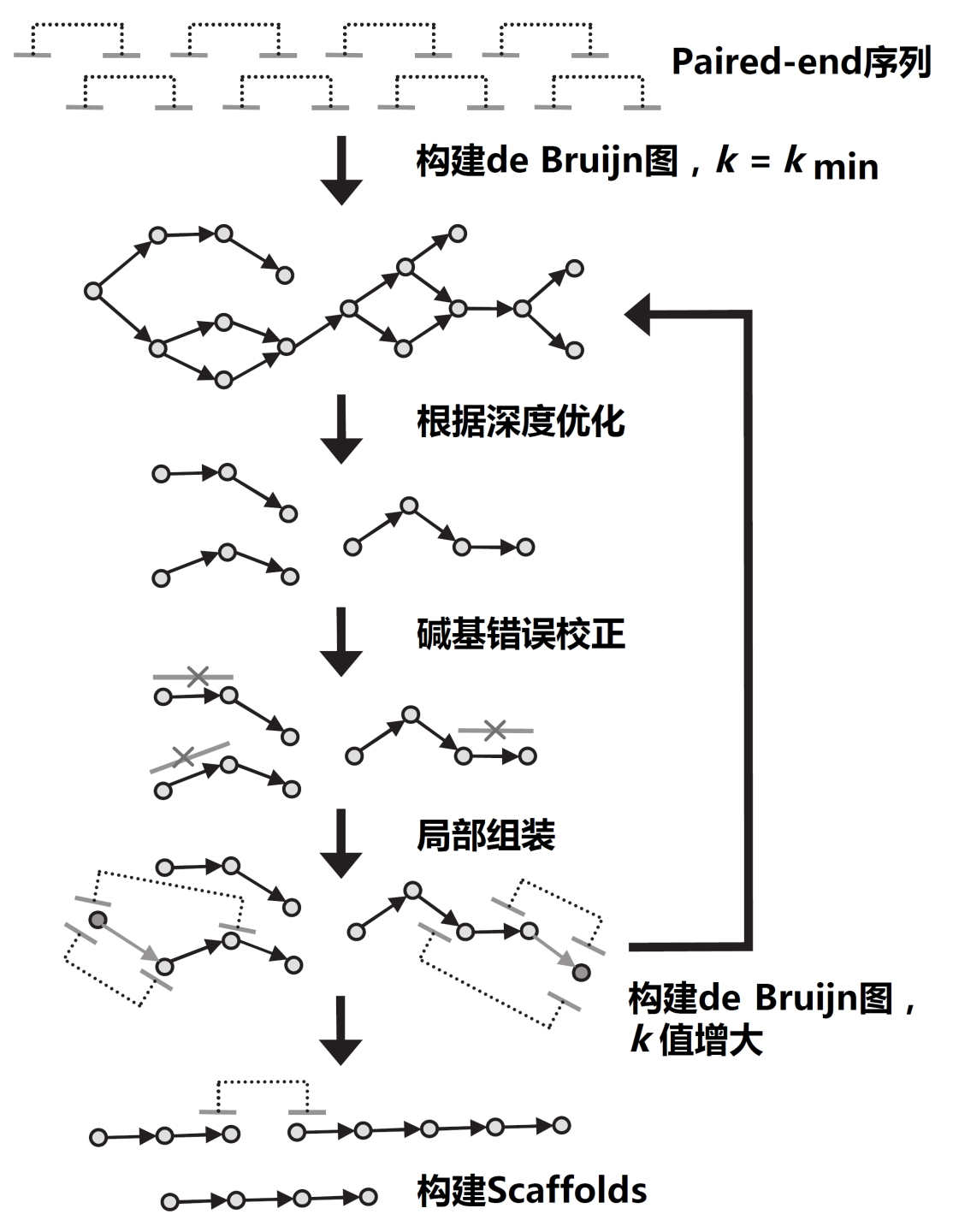
IDBA-UD拼接組裝流程圖,修改自文獻(xiàn)[4]
當(dāng)然,de Bruijn圖也并非萬能。對于最近日漸流行的三代單分子實時測序技術(shù),由于其具有超長讀長的特性,HGAP(Hierarchical Genome Assembly Process)[5]等根據(jù)序列比對尋找彼此之間重疊區(qū)域的方法更為合理。
4. 拼接效果的評價
正如游戲得分有高低,在拼接完成后,我們也需要對組裝效果進(jìn)行評估。顯然,Contigs和Scaffolds長度是評價的重要標(biāo)準(zhǔn)之一。通常我們使用N50值來評估,將所有Contigs/Scaffolds序列按照長度從長到短依次排列后相加,當(dāng)加和的長度達(dá)到總長度的50%時,最后一條Contigs/Scaffolds序列的對應(yīng)長度即N50值。顯然N50越長,拼接組裝效果越好,宏基因組序列也就越完整。
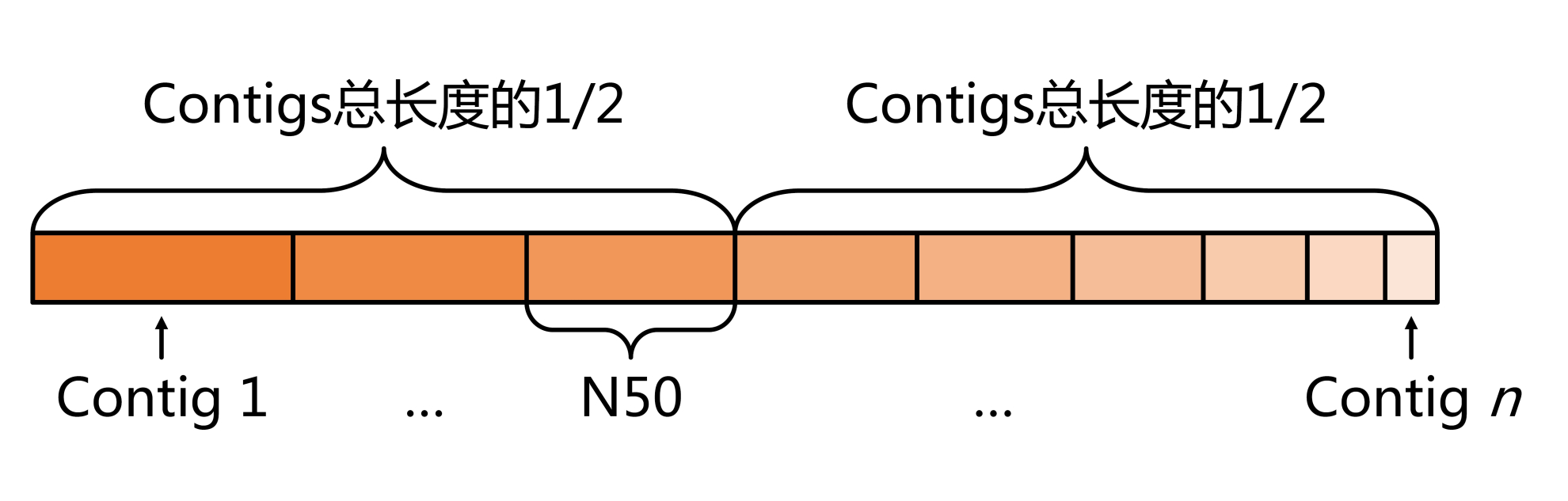
N50值計算示意圖
當(dāng)然,宏基因組的復(fù)雜程度將直接影響拼接組裝的效果。比如,對于腸道宏基因組樣本,迄今為止的幾項大型研究(如MetaHIT、HMP和IGC等項目)得到的基因目錄的N50值都在1 kb左右[6-8]。
結(jié)語
通過以上的講解,小伙伴們對宏基因組的“拼圖游戲”應(yīng)該入門了吧!總體而言,拼圖游戲雖然復(fù)雜,但只要選對合適的算法,通關(guān)也并非遙不可及!當(dāng)然,隨著科技的不斷發(fā)展,我們也期待涌現(xiàn)更多更強大的宏基因組拼接組裝工具。
至于拼接獲得的Contigs/Scaffolds序列如何用于后續(xù)分析,且待下回分解,敬請各位小伙伴保持關(guān)注哦!
附:【5分鐘小課堂】后續(xù)預(yù)告
l 看不見摸不著的它們,都在忙些啥?宏基因組功能注釋為您解答!
l 茫茫菌群,誰是天使,誰是元兇,誰又是圍觀路人甲?
l 菌株水平的超高分辨率解析,宏基因組學(xué)就是這么高大上!
參考文獻(xiàn)
1. Fan W, Li RQ (2012) Test driving genome assemblers. Nature Biotechnology 30: 330-331.
2. Berger B, Peng J, Singh M (2013) Computational solutions for omics data. Nature Reviews Genetics 14: 333-346.
3. Luo RB, Liu BH, Xie YL, Li ZY, Huang WH, et al. (2012) SOAPdenovo2: an empirically improved memory-efficient short-read de novo assembler. GigaScience 1: 6.
4. Peng Y, Leung HCM, Yiu SM, Chin FYL (2012) IDBA-UD: a de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 28: 1420-1428.
5. Chin CS, Alexander DH, Marks P, Klammer AA, Drake J, et al. (2013) Nonhybrid, finished microbial genome assemblies from long-read SMRT sequencing data. Nat Methods 10: 563-569.
6. Huttenhower C, Gevers D, Knight R, Abubucker S, Badger JH, et al. (2012) Structure, function and diversity of the healthy human microbiome. Nature 486: 207-214.
7. Li JH, Jia HJ, Cai XH, Zhong HZ, Feng Q, et al. (2014) An integrated catalog of reference genes in the human gut microbiome. Nature Biotechnology 32: 834-841.
8. Qin JJ, Li RQ, Raes J, Arumugam M, Burgdorf KS, et al. (2010) A human gut microbial gene catalogue established by metagenomic sequencing. Nature 464: 59-65.




















