

2017-02-17
簡化基因組測序(Reduced-Representation Genome Sequencing, RRGS)是指利用生物信息學方法,設計分子標記開發方案,篩選特異性長度片段,應用高通量測序技術獲得海量標簽序列來充分代表目標物種全基因組信息的測序策略。簡言之,簡化基因組測序就是基因組重測序的簡化版,小編稱之為 “省錢的重測序”。
目前,主流的簡化基因組建庫方法包括 RAD 法(Restriction site Associated DNA),2b-RAD 法、GBS 法(Genotyping-By-Sequencing)、ddRAD 法(double-digest RAD,由于該種方法建庫的流程更加類似于 GBS 法,所以也被稱為 ddGBS 法)。今天,小編就給大家簡要地介紹一下 GBS 分析的一般流程。
GBS分析流程的三個主要步驟:
1. 原始數據處理
原始數據通常包含各種類型的錯誤,如堿基錯配、低質量堿基、接頭污染。因此,在進行后續分析前,小編強烈建議對下機數據進行質量評估、接頭序列去除、低質量數據過濾。目前主流的用于原始數據處理的軟件包括:Trimmomatic、PRINSFQ、FastqMcf、FASTX-Toolkit和AdapterRemove2等。
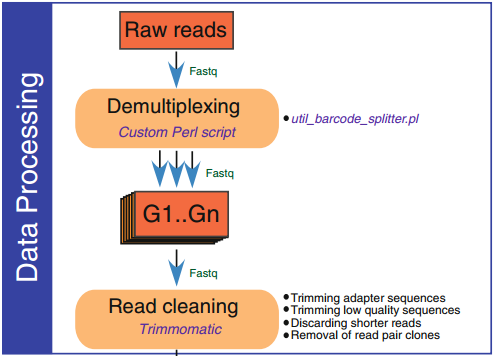
2. 比對到參考基因組
數據過濾后,對于有參考基因組的物種,我們可以將短讀長比對到參考基因組,序列比對軟件包括(MAP、mrsFast、STAMPY、Bowtie2、BWA和SOAP2等)。對于無參考基因組的物種,我們可以利用STACKS,UNEAK或者RApiD等軟件將測序得到的 reads 拼接成 mini-contigs。這些mini-contigs可用于短讀長序列比對和基因分型。
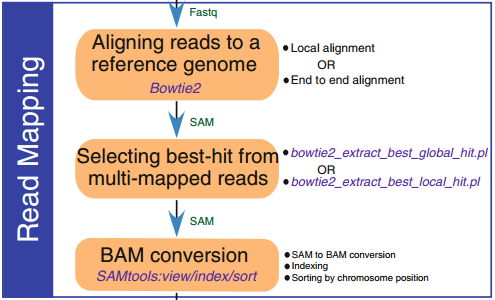
3. 變異檢測
比對到參考基因組后,一般會生成BAM文件。然后就可以檢測個體或群體的變異(包括SNPs和InDels)。用于變異檢測的軟件有SAMtools、GATK,SOAP,SNVer和GNUMAP等。
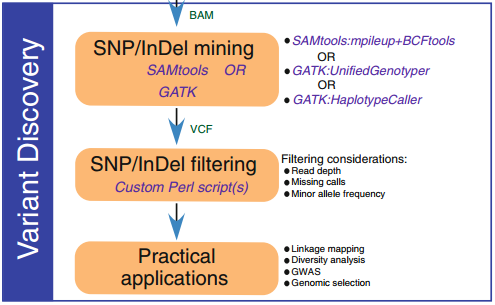
小結:
本文簡單介紹了GBS分析的一般流程,以及在每一個流程中需要用到的一些軟件。本流程中選擇的軟件均為運算效率較高的一些軟件,具體軟件見表1。
表1 GBS 分析流程中使用的軟件

此外,小編還想給想要做 GBS或正在做 GBS 的老師們一些建議:
(1) 測序數據量很重要!很重要!!很重要!!!數據量 = 標簽數*測序深度*讀長;
(數據量太少,樣品之間測到的標簽會不在基因組的同一位置)
(2) 標簽數不是隨意定的,酶切位點決定標簽數量,所以選擇一款合適的酶很重要;
(3) 實驗過程很關鍵,PCR 循環可能會引入大量的重復序列,所以在建庫過程中,一定要降低 PCR 循環數!降低PCR 循環數!!降低PCR 循環數!!!
(重復序列的存在會影響變異的檢測)
原文索引: DataKagale S , Koh C , Clarke WE. Analysis of Genotyping-by-Sequencing (GBS)[J]Methods in Molecular Biology, 2016


















