

2017-03-01

該研究于2016年發表于Nature子刊《Scientific Reports》上
(最新影響因子:5.228)
研究背景:
拼接組裝一直以來都是宏基因組項目中最重要的一個環節,它對后續的基因功能注釋、微生物基因組重建以及物種分類注釋都起到很大的影響。目前,Illumina平臺較短的測序片段對宏基因組拼接結果影響較大,而三代基于PacBio的SMRT單分子實時測序技術的興起,將有望顯著提升宏基因拼接組裝的效果。
研究方法:
樣本來源:沼氣反應池微生物群落
測序平臺:PacBio RS II+Illumina HiSeq
通過Hiseq和PacBio平臺的測序分析,比較兩個平臺的宏基因組組裝的效果以及物種注釋精確度。
研究結果:
Hiseq平臺產出18.5 Gb宏基因組數據用于拼接,共拼接得到3,035,577個Contigs,平均189 nt,55,633個Contigs> 1 kb,最大長度148,797 nt。而利用PacBio平臺產出的95.4 Mb數據用于拼接,共拼接出2,181 個contigs,平均長度4,459 nt,最大長度65,165 nt。
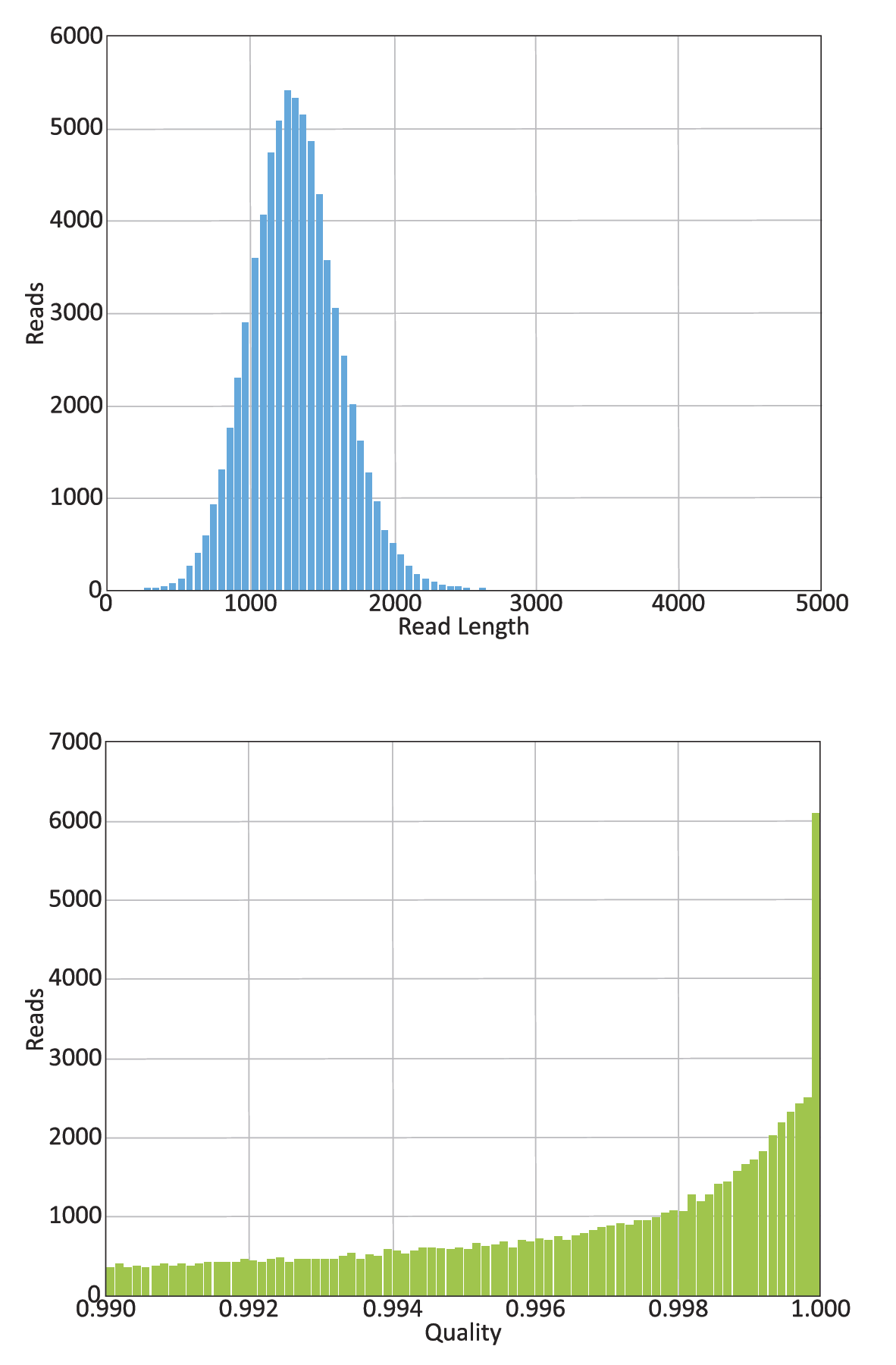
采用PacBio RS II測序平臺中的P4-C2試劑盒,插入1.5kb片段構建文庫進行CCS測序,在獲得的測序結果中,上圖為質量分數高于99%的序列長度分布統計以及序列質量分布統計,其中共71,254條序列質量高于99%,平均序列質量高達99.7%。
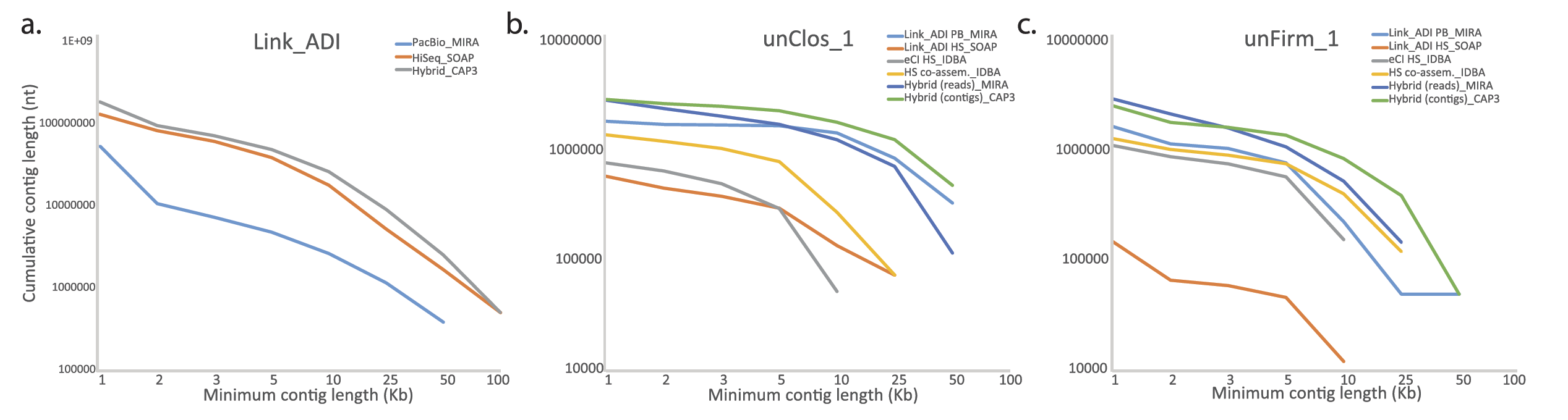
不同測序平臺、不同拼接軟件對宏基因組測序數據組裝拼接獲得的Contig長度比較。
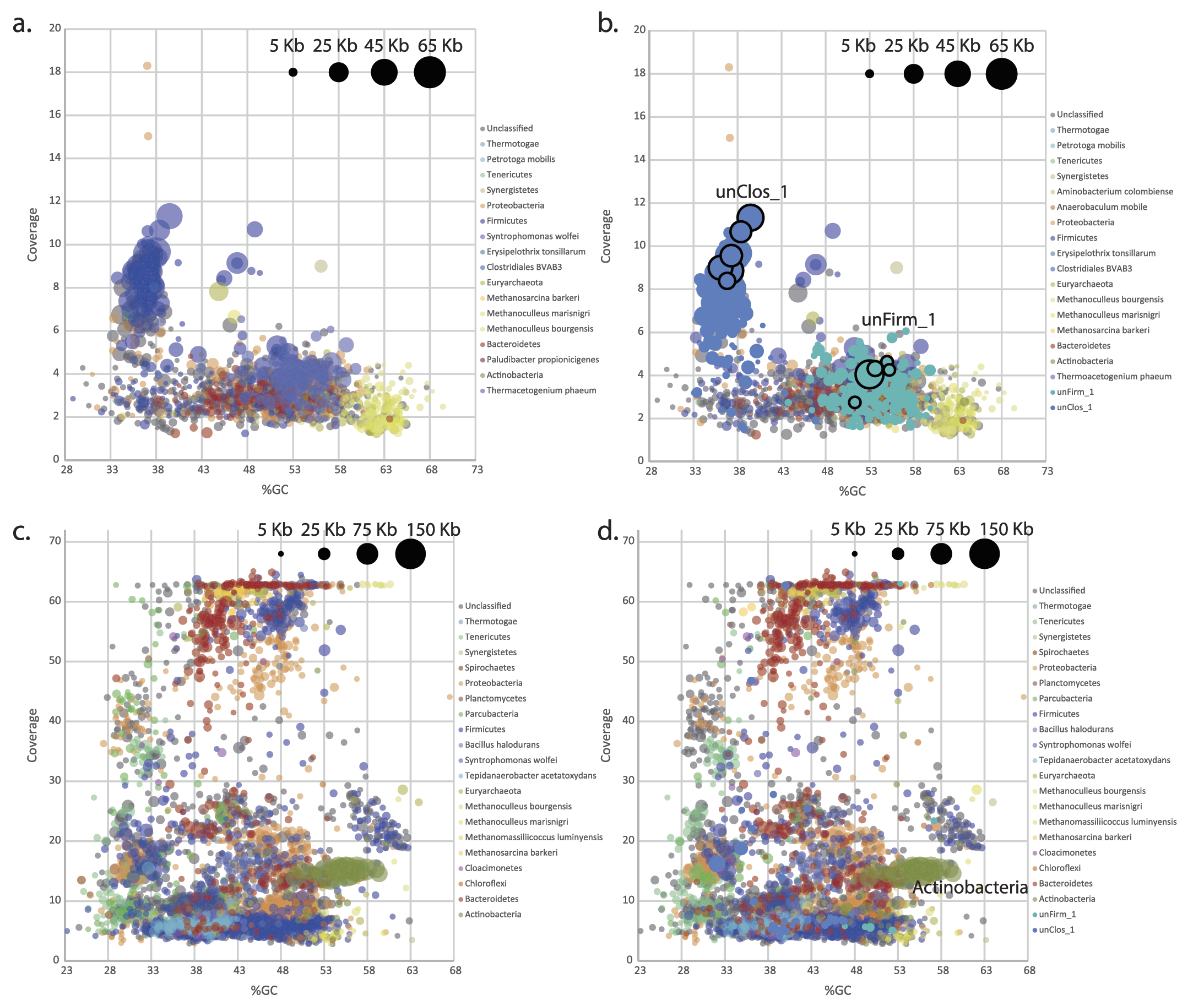
對同一份樣本分別采用PacBio(a-b)和HiSeq(c-d)測序,運用PhyloPythiaS進行物種分類注釋,并對注釋結果(GC含量、覆蓋度和Contig長度)進行可視化比較。由圖可見,三代PacBio平臺獲得的Contigs更長更完整,二代HiSeq平臺獲得的Contig碎片多得多。b圖和d圖在注釋分析時加入了物種特異性的訓練數據集。
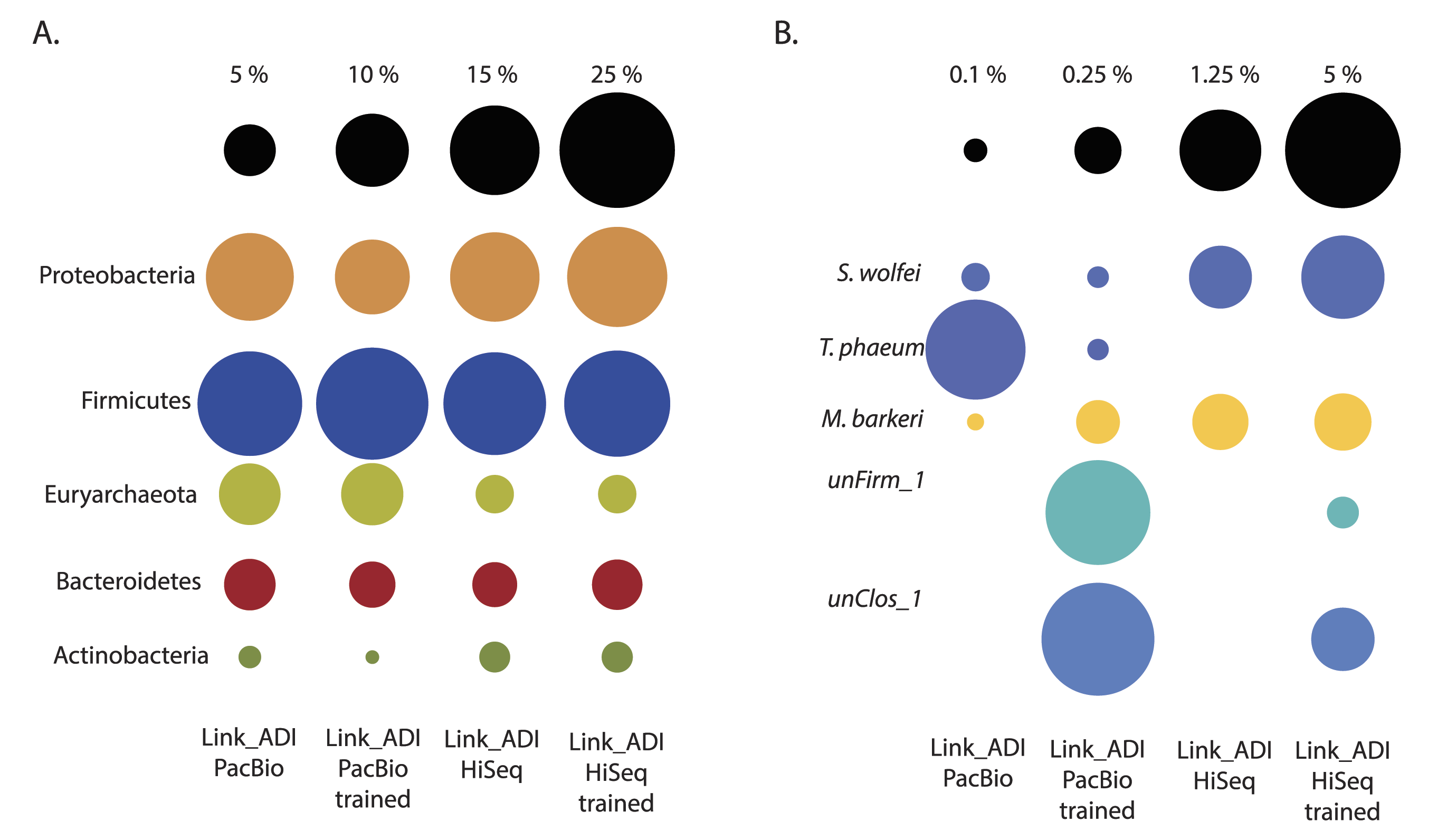
對兩種不同平臺物種分類注釋結果的比較,分別基于(A)圖門水平與(B)圖種水平的物種豐度比較結果。由圖可知,在門水平,兩個平臺測序數據的物種豐度較為一致,但在種水平,不同物種的豐度差異比較明顯,尤其在加入了物種特異性的訓練數據集后,PacBio平臺對于unFirm_1與unClos_1兩個物種的豐度結果統計更加精準。
研究結論:
綜上所述,運用三代PacBio測序技術,將顯著提升宏基因組數據的拼接效率和物種注釋精準度,相比二代Illumina HiSeq測序平臺,可謂是取得了質的飛躍!
派森諾優勢
2016年,派森諾生物在原有的PacBio RS II三代高通量測序儀基礎上,率先部署最新款Sequel測序儀,并已投入使用,獨家提供三代測序分析服務,助力微生物組研究!
作為行業先鋒,派森諾生物將一如既往地行使“解析序列,詮釋生命”的理念,秉承“立足客戶需要,滿足個性需求”的服務宗旨,始終如一地提供性價比最高、最優質、最快速穩定的高通量測序和數據解析方案。
派森諾生物將竭誠為您服務!
參考文獻
Frank, J. A. et al. Improved metagenome assemblies and taxonomic binning using longread
circular consensus sequence data. Sci. Rep. 6, 25373; doi: 10.1038/srep25373 (2016).


















