

2017-03-01
以PacBio公司的SMRT單分子實時測序技術(Single molecule real-time sequencing)為代表的三代測序技術,通過其獨有的環形一致性測序模式(Circular-consensus sequence,CCS),極大提高單堿基測序的準確率,遠超Illumina等二代測序技術。與傳統轉錄組測序項目相比,利用PacBio平臺的全長轉錄組測序技術可以直接獲得mRNA的全長,保證了mRNA序列的精確性。近期我們將陸續推出全長轉錄組測序技術相關文章,供討論和交流。我們在上一期向大家介紹了全長轉錄組測序的技術原理和實驗流程,本期將為大家介紹全長轉錄組測序分析流程中的數據整理和質控。
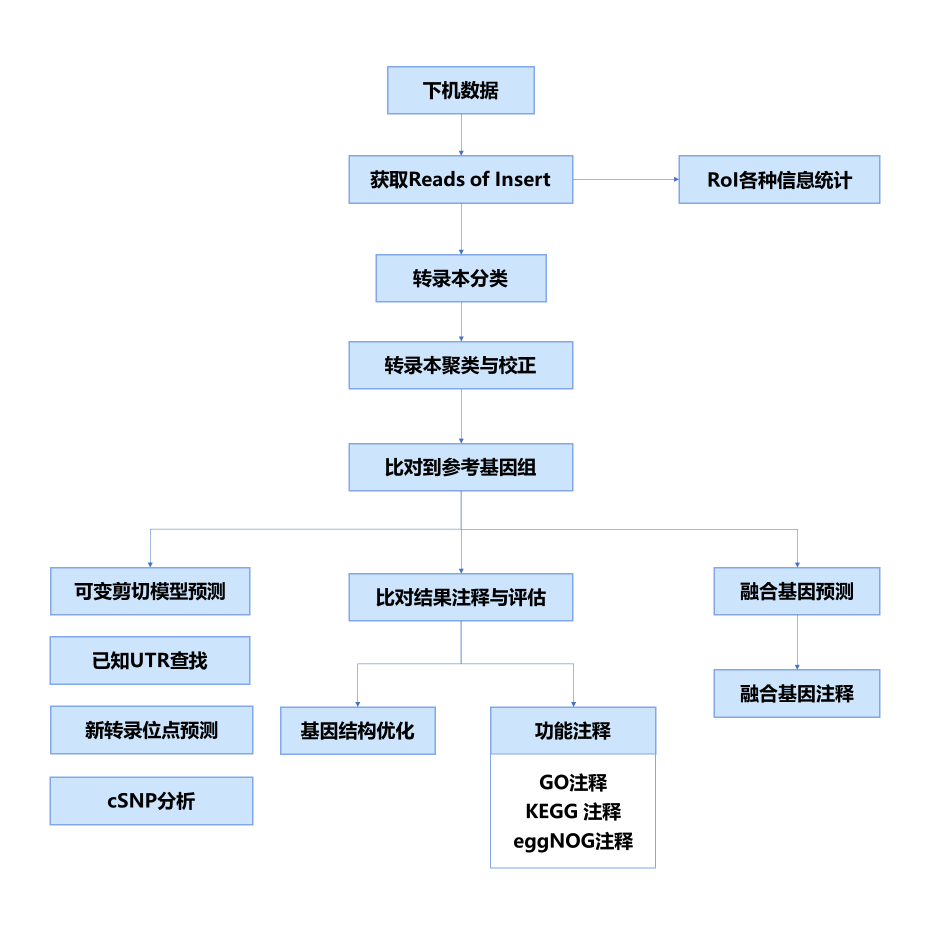
分析流程
首先從下機數據中提取Reads of Insert(RoI)序列,根據Reads of Insert序列是否含3’ 端引物和5’端引物以及是否嵌合對Reads of Insert進行分類,對全長序列進行去冗余聚類,并用非全長序列對其進行校正。接著將非冗余序列比對到該物種的參考基因組上,在此基礎上完成融合基因分析,并進行比對結果整理,根據比對結果與參考基因組的注釋信息對Isoform進行功能注釋,并完成基因結構優化。另外,我們還進行了轉錄本結構分析,包括可變剪接分析, UTR區域注釋,cSNP 和InDel 分析等。
數據整理和質控
每個原始序列可以分割成一個或多個子序列(Subread,Subread 是DNA聚合酶以一條模板鏈經過一個Passes合成的,不包括Adapter序列),即每個零模波導孔中會有多個Subreads。每個零模波導孔中的所有Subreads來自同一個轉錄本,由于其堿基出錯率是隨機的,可通過Subreads間比對提高堿基質量,獲得一條Reads of Insert,即Reads of Insert通過同一零模波導孔中的Subreads校正后得到。
對原始下機數據進行提取和過濾Subreads,去除Adapter和低質量的序列。為了充分利用數據,篩選出長度大于50bp、序列準確度大于0.8并且Full Passes數目大于0的序列,得到Reads of Insert,如圖1。一個Full passes指原始序列中的一條子序列兩端均含有 Adapter(圖中黑色區域),一個原始序列的Full passes數目指在該序列中文庫cDNA序列被完整測到的次數,圖1中有Full passes數目為2。

圖1 Reads of Insert示意圖
我們對每個樣品的Reads of Insert按不同插入片段長度分別進行統計,包括RoI序列數目、RoI總堿基量、RoI序列平均長度、RoI序列平均質量和平均Passes。整理好的數據就可以開展后續的分析了,具體分析內容將在下一期進行闡述,敬請期待。


















