

2017-03-08
以PacBio公司的SMRT單分子實時測序技術(Single molecule real-time sequencing)為代表的三代測序技術,通過其獨有的環形一致性測序模式(Circular-consensus sequence,CCS),極大提高了單堿基測序的準確率,遠超Illumina等二代測序技術。與傳統轉錄組測序項目相比,利用PacBio平臺的全長轉錄組測序技術可以直接獲得mRNA的全長,保證了mRNA序列的精確性。上期我們為大家介紹了全長轉錄組測序的數據質控,本期將為大家介紹三代全長有參轉錄組的分析內容。主要包括:
1. 轉錄本分類
數據過濾后得到高質量的Reads of Insert(RoI),接著對其進行轉錄本分類。理論上完整的RoI應該有5’primer、3’primer和polyA部分。但在實際測序中,由于5’端降解等因素,并不是所有的RoI都是完整的,即不是全長轉錄本序列,而且有少部分RoI序列為嵌合體。因此通過檢測RoI序列是否含有 5’primer, 3’primer和polyA以及其位置的關系, 將RoI序列分為全長非嵌合(Full-Length-Non-Chimeric)序列、全長嵌合(Full-Length-Chimeric)序列、非全長(Non-Full-Length)序列等。其中全長非嵌合序列即是物種原始的全長mRNA序列,各分類之間的相互關系見下圖:
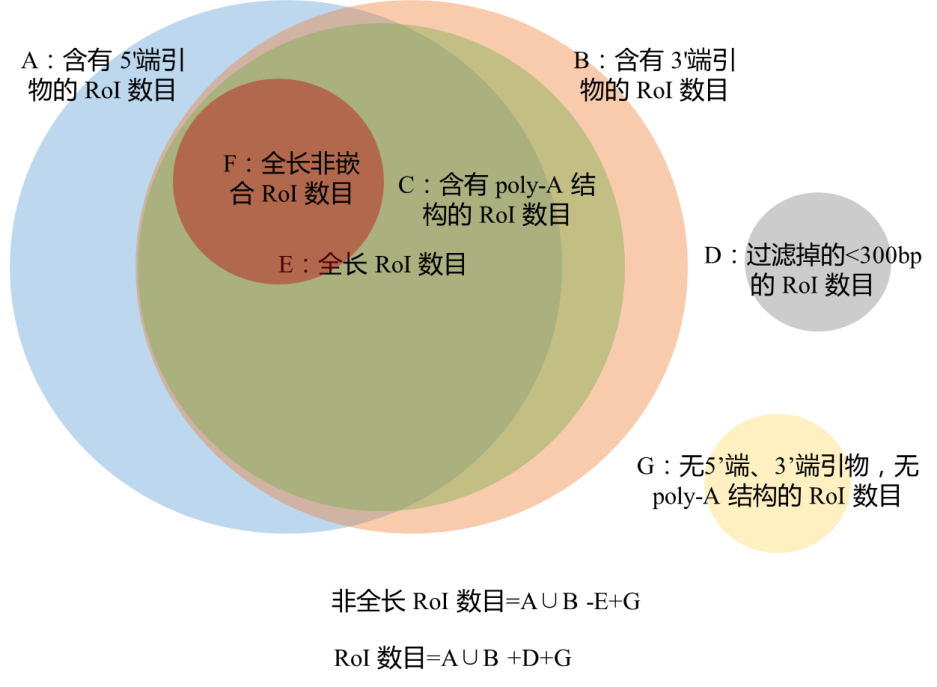
圖1 轉錄本分類
2. 序列聚類與矯正
全長非嵌合序列中存在大量的冗余序列,我們將冗余序列聚類到一起,得到新的一致性序列(Consensus Isoforms),然后將非全長序列比對到一致性序列上進行校正,最終得到準確度大于99% 的高質量Isoform(即轉錄本)。下圖是Isoform長度分布圖,長度分布的區間與文庫片段的選擇有關。
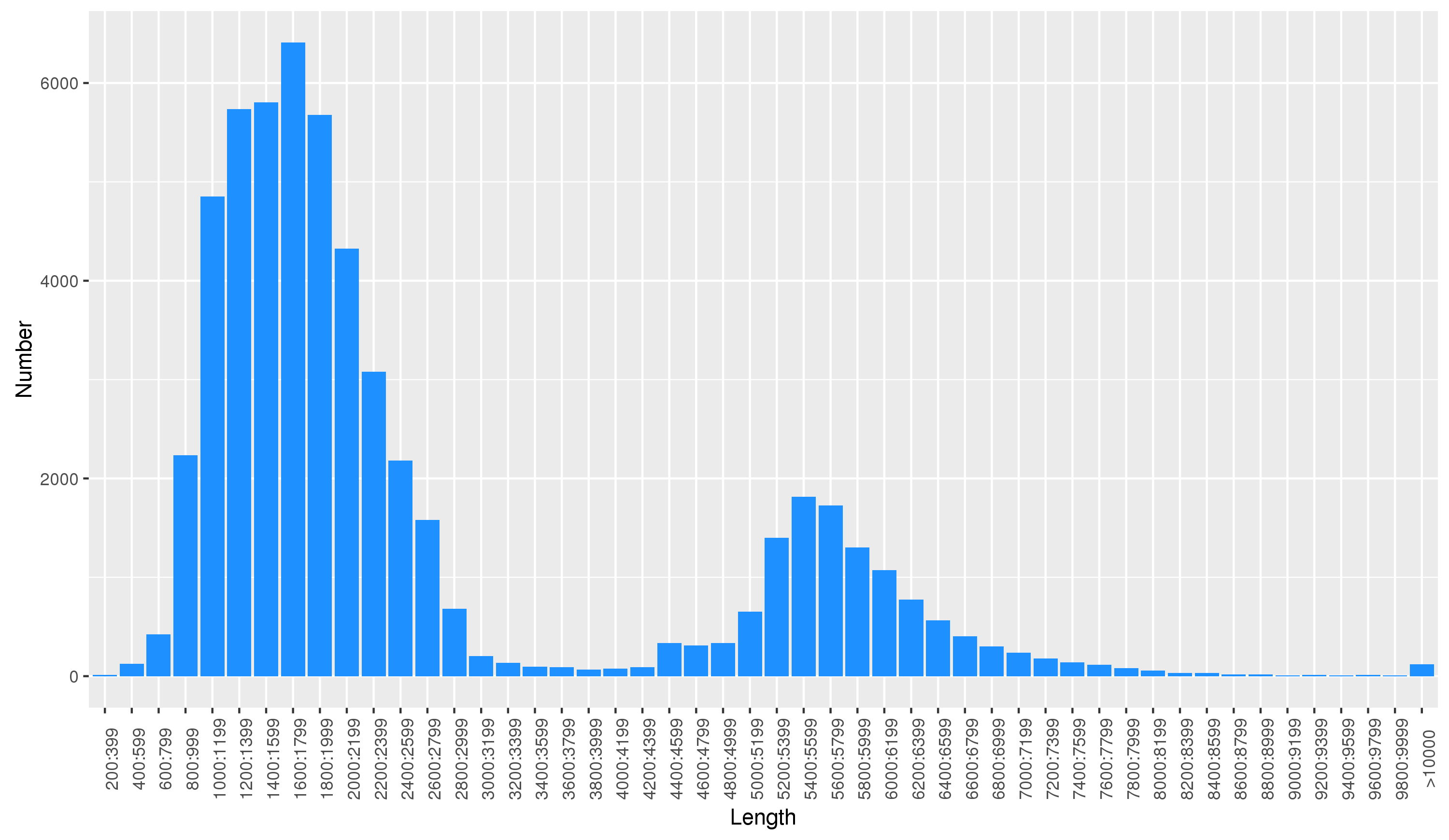
圖2 Isoform長度分布
3. 參考基因組比對
我們將經過校正得到的Isoform與參考基因組比對, 根據基因組注釋信息,可得知序列的來源基因以及表達產物的結構。與參考基因組比對情況見下圖,圖中Score 0:覆蓋了基因的區域,但是沒有或者少有與基因的exon匹配上的Isoform數目;Score 1:與基因的一些exon有一對一overlap的Isoform數目; Score 2:與基因的一些exon有一對一overlap并且匹配情況較好的Isoform數目。
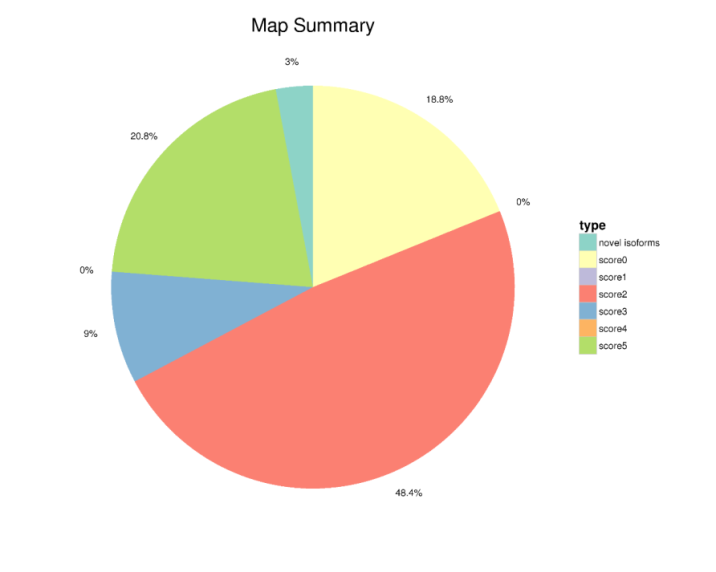
圖3 比對參考基因組情況統計
4. Known Isoforms 和Novel Isoforms功能注釋
對于比對到基因組上的score為0-5的known Isoform采用有參考基因組注釋方法,對于Novel Isoform采用無參考基因組注釋方法。基因功能注釋所用到的數據庫包括 GO、KEGG、eggNOG、NR、Swiss-Prot等。下圖是其中的eggNOG注釋結果:

圖4 eggNOG注釋
5. 結構分析
結構分析是三代全長轉錄組中的一個重點研究內容,不同的樣本轉錄物不盡相同,我們進行轉錄本結構分析,可以統計所有轉錄本的結構差異。它能夠準確辨別二代測序無法識別的同源異構體或同源基因等。轉錄本結構分析包括可變剪接,融合基因, UTR區域注釋,cSNP 和InDel 分析等。
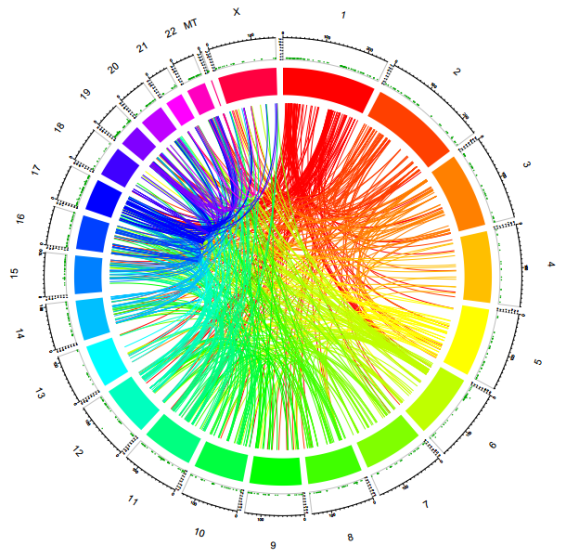
圖5 融合基因分析
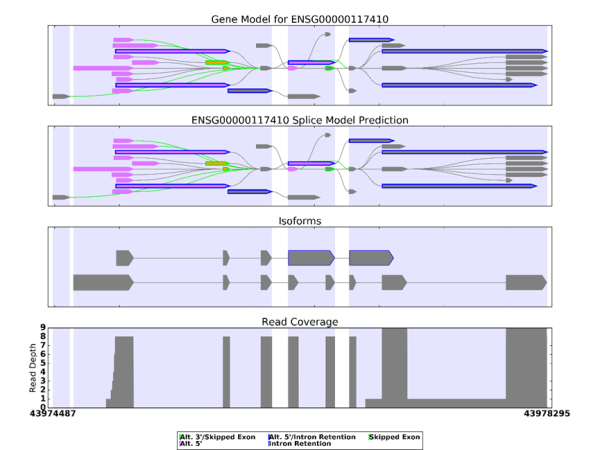
圖6 可變剪切分析


















